Relazione di tirocinio
Fallini
Riccardo
Dal 31 Agosto al 30 Settembre 1998, ho svolto
un’attività di tirocinio al Dipartimento di Farmacologia
dell’Università Statale di Milano, nel laboratorio del Dott.
Torsello.
Il laboratorio seguiva diverse linee di ricerca riguardanti neoplasie,
ormoni, etc. Io mi sono occupato dell’ormone della crescita
(GH). La ricerca era orientata a trovare una metodica relativa
all’analisi quantitativa dell’ormone nei tessuti ipofisari
e ipotalamici, in condizioni fisiologiche.
Quest’ormone è presente in quantità talmente basse (picogrammi:
pg =10-9g) da non essere rilevabile con i normali metodi
d’analisi (HPLC, etc.). Pertanto la ricerca aveva come obiettivo
l’isolamento e la quantificazione dell’mRNA del GH.
Per fare ciò abbiamo una tecnica detta RT-PCR (RT: retrotrascrizione;
PCR: reazione di polimerizzazione a catena).
Essa prevede:
- Estrazione RNA totale dal tessuto
- Aggiunta, a diverse concentrazioni, a diversi
campioni, di DNA sintetizzato in questo modo:
- Aggiunta di 300 paia di basi (pb, cioè
il numero di nucleotidi appaiati nel doppio filamento di DNA)
nella parte centrale della sequenza del GH, le paia di estremità
terminali sono, infatti, quelle decisive per il proseguimento
della tecnica.
Questa sequenza nucleotidica ha la funzione
di competere con l’RNA corrispondente (è detto infatti RNA
competitivo).
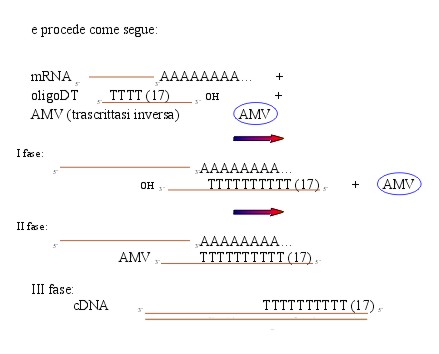
3. RT, cioè sintesi del DNA codificante (cDNA)
dall’mRNA.
Il meccanismo richiede l’aggiunta
di:
I fase: appaiamento degli oligoDT agli mRNA,
l’oligoDT, dato il numero elevato di timine, è complementare
solo con gli mRNA, poiché la presenza di una coda di adenine,
complementari alle timine, è una caratteristica di tutti gli
mRNA.
II fase: l’oligoDT fornisce l’OH iniziale per la trascrittasi
inversa che può quindi trasformare l’mRNA (l’unico
che ha l’oligoDT appaiato) in DNA (cDNA). All’RNA
competitivo non succede niente perché non ha una zona di appaiamento
e perché è già DNA (III fase).
4. PCR, cioè amplificazione del DNA (sia l’RNA
competitivo sia il cDNA). Essa prevede l’aggiunta di:
- 2 primers (complementari alla parte iniziale
del gene, uno in direzione 5’-3’, l’altro 3’-5’;
essi forniscono l’OH iniziale per la DNA polimerasi)
- enzima DNA polimerasi (TAQ, estratta da un termofilo per non
degradarsi alle alte temperature della tecnica)
e procede come segue:
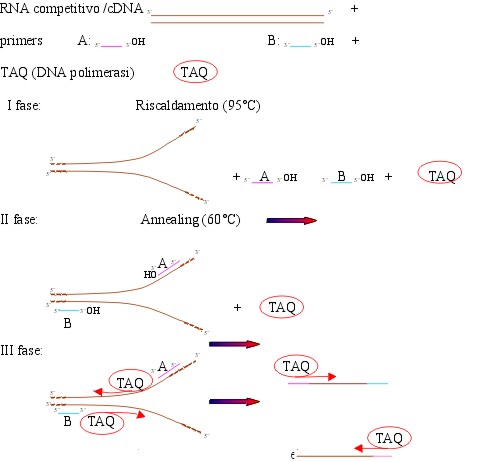
I fase: riscaldamento a 95°C : si ha così
la denaturazione dei frammenti di DNA, cioè l’apertura
del doppio filamento
II fase: annealing, cioè appaiamento dei primers alla zona di
complementarità , essi si appaiano sia al cDNA sia all’RNA
competitivo
III fase: l’enzima si attacca ai primers incominciando
a replicare la sequenza di DNA a valle dei primers. Si ripete
il ciclo per 25 volte.
Nei campioni in cui si hanno alte concentrazioni
di RNA competitivo, si ha una maggiore amplificazione di questo,
rispetto a quella che avviene nei campioni con basse concentrazioni
di RNA competitivo
5. Valutazione della riuscita dell’esperimento:
ai campioni ottenuti aggiungo:
- Etidio di Bromuro (EtBr), è una sostanza
fosforescente agli UV e intercalandosi (cioè si posiziona tra
le basi) mi fornisce una luminosità proporzionale alla quantità
di DNA presente nel campione.
Faccio un’elettroforesi su agarosio dei diversi campioni (preparati
nel punto 2). Se si verifica una inversa proporzionalità tra le quantità
di DNA (il cDNA originato dall’mRNA e l’RNA competitivo),
posso affermare che quando il rapporto tra la luminosità (proporzionale
alla concentrazione) dell’mRNA e quello dell’RNA competitivo
è uguale a 0.5, la concentrazione dell’mRNA è uguale a quella
dell’RNA competitivo. Conoscendo la concentrazione di quest’ultimo,
posso allora determinare la concentrazione dell’mRNA.
Questa tecnica in realtà non è stata realizzata,
perché sono insorte delle difficoltà nella costruzione dell’RNA
competitivo, per il seguente motivo.
Non erano presenti siti di restrizione (e di ligasi) comuni e
unici tra la sequenza nucleotidica del GH e quella del segmento
di DNA da aggiungere (300pb del gene della prolattina) per la
sintesi dell’RNA competitivo (forse si sarebbe potuto scegliere
un altro pezzo anche di un altro gene, per trovare le condizioni
richieste).
Comunque in seguito ad una serie di tentativi (non riusciti) con
enzimi di restrizione aventi più siti di taglio, ho avuto la possibilità
di applicare processi tipici dell’ingegneria genetica quali:
- La trasformazione di cellule batteriche,
le quali assumono, grazie a questo processo, capacità normalmente
non possedute come, ad esempio, la sintesi di una certa proteina.
- L’amplificazione di un plasmide: ottenere,
cioè, innumerevoli copie di quel pezzo di DNA circolare.
- Ingegnerizzare un plasmide, cioè togliere
e aggiungere sequenze (che possono essere geni) ad un plasmide.
- L’elettroforesi su agarosio di DNA,
tecnica che permette di distinguere sequenze di DNA o RNA, aventi
lunghezze diverse, potendola anche valutare .
Nello stesso istituto ho imparato tecniche
come l’uso di:
- HPLC, che permette l’analisi quantitativa
di diverse proteine presenti in un liquido corporeo o meno,
come plasma o un estratto tessutale
- Termociclatori, per far compiere al campione
un numero di cicli costituiti da temperature e tempi diversi.
Sono utilizzati per la RT e per la PCR
- Transluminatori agli UV, per rendere visibili
i campioni nell’elettroforesi di DNA o RNA
- Tecniche di separazione proteica: Western
blot e ligand
Ho acquistato anche una certa manualità con
microscopi elettronici a scansione e a trasmissione, microtomi
e ultramicrotomi (per la preparazione di campioni visibili al
microscopio elettronico), e spettrofotometri UV-visibili, utilizzato
per determinare la concentrazione degli acidi nucleici
|
|