| Autore |
Discussione |
|
|
weppa29
Nuovo Arrivato

6 Messaggi |
 Inserito il - 09 maggio 2019 : 18:45:31 Inserito il - 09 maggio 2019 : 18:45:31



|
Buonasera a tutti, sono lieto di far parte di questo forum.
Sto occupandomi di clonaggio genico e vorrei esporvi quanto accadutomi qualche giorno fa.
Ho purificato con PlasmidPrep Mini Spin Kit i miei cloni di pJET 1.2 e li ho sottoposti a digestione con differenti enzimi affinch� liberassero gli inserti (avrei dovuto clonarli in un secondo vettore), utilizzando:
- 12 microlitri di acqua;
- 5 microlitri di purificazione;
- 2 microlitri di buffer (Buffer 3.1 per BglII e Buffer FastDigest per KpnI e BamHI);
- 1 microlitro di enzima (BglII, BamHI, KpnI).
Allo stesso modo, ho digerito il secondo vettore nel quale avrei dovuto clonare i miei frammenti, utilizzando per� solo BamHI e KpnI (i siti di restrizione di BamHI e BglII sono compatibili).
In entrambi i casi, la digestione � avvenuta a 37�C per quasi un'ora.
Al termine della digestione, ho effettuato una gel elettroforesi, caricando 5 microlitri di campione purificato e 20 microlitri di campione digerito (il mio obiettivo era quello di purificare l'inserto da gel e clonare nel nuovo vettore).
Per quanto riguarda il vettore, per verificare l'avvenuta digestione ho caricato su gel di agarosio 5 microlitri di vettore purificato ma non digerito, 5 microlitri di vettore digerito da KpnI e 5 microlitri di vettore digerito da BamHI.
Il risultato � il seguente (foto 1, digestione cloni, foto 2 digestione vettore)...nessuna banda laddove ho caricato il clone digerito (ho ottenuto solo due bande nell'ultimo campione, quella di 3000 bp dovrebbe essere del vettore e quella di circa 2000 dell'inserto)e banda molto debole quando ho digerito il vettore con BamHI. Ho rifatto tutto aumentando la quantit� di DNA (sia del vettore che dei cloni), ma non ho ottenuto dei cambiamenti significativi.
Secondo voi quale potrebbe essere il problema? Grazie!
Immagine:
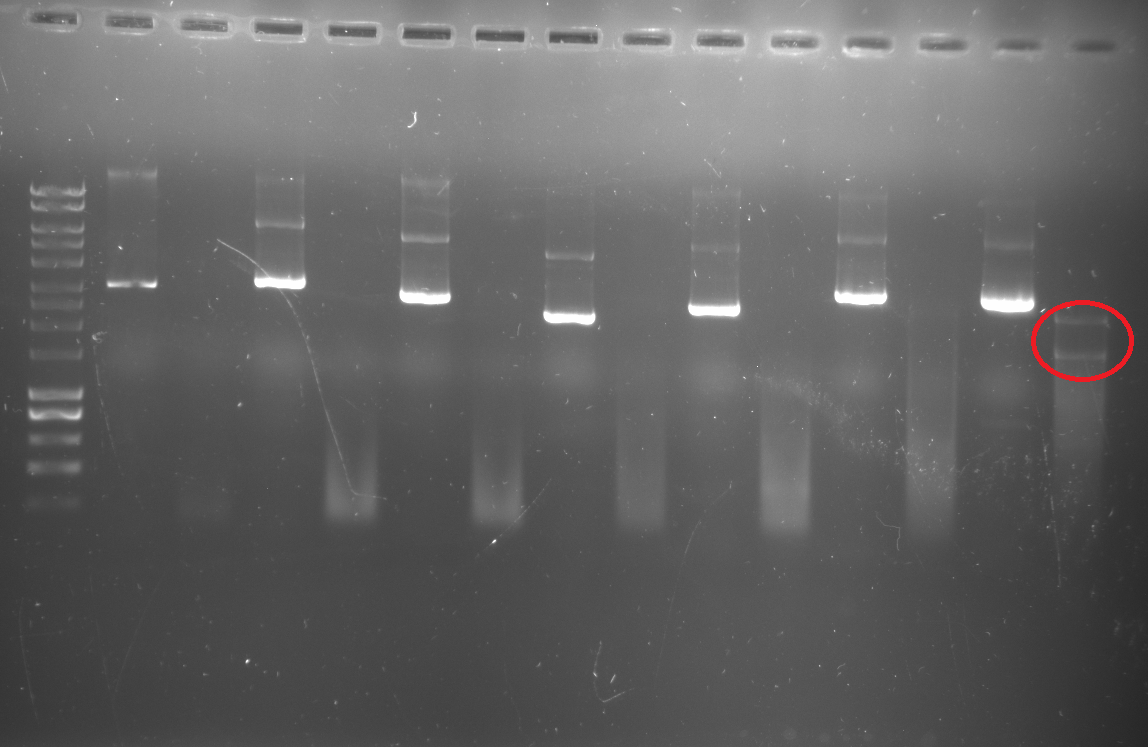
788886
Immagine:
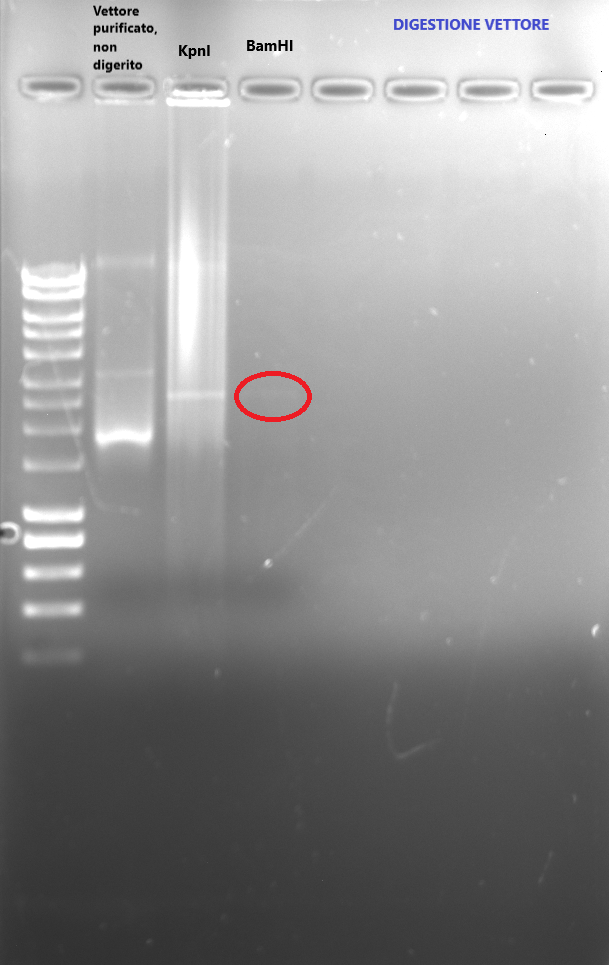
482926
|
|
|
|
|
weppa29
Nuovo Arrivato
6 Messaggi |
Inserito il - 12 maggio 2019 : 12:15:12
|
  |
 |
|
|
domi84
Moderatore


Citt�: Glasgow
1724 Messaggi |
|
|
weppa29
Nuovo Arrivato
6 Messaggi |
|
|
Geeko
Utente

Citt�: Milano
1043 Messaggi |
Inserito il - 13 maggio 2019 : 21:58:28

|
| A mio parere hai un problema di degradazione (e anche un po' di contaminazione da DNA genomico). La degradazione pu� derivare principalmente da due fonti: (1) la quantit� eccessiva di enzimi di restrizioni che stai usando. Vero che di solito se ne usi troppo puoi incorrere in prodotti di digestioni non specifici, e non in una completa degradazione. Prova a usare sulle 5U di enzima per microgrammo di DNA, non tanto di pi�. (2) la spiegazione pi� plausibile � che tu abbia una contaminazione da DNasi nelle soluzioni che stai usando, verosimilmente l'acqua stessa. Preparati tampone di corsa fresco fatto in acqua mq (meglio ancora se autoclavata) e usa lo stesso per fare il gel. I plasmidi integri sono resistenti alle esonucleasi, ma non appena li digerisci non lo sono pi�. Facci sapere. |

|
|
|
|
weppa29
Nuovo Arrivato
6 Messaggi |
 Inserito il - 14 maggio 2019 : 08:51:27 Inserito il - 14 maggio 2019 : 08:51:27
|
 Scritto da MolecularLab Mobile Scritto da MolecularLab Mobile
Grazie mille! Una contaminazione da DNasi di puntali e pipette suppongo sia meno probabile, giusto? Credo che il problema non siano neanche i buffer (ne ho usati due differenti e mi sembra strano che fossero entrambi contaminati).
Per quanto riguarda la contaminazione da DNA genomica? Da cosa si deduce che sia avvenuta? Grazie ancora per il prezioso aiuto :) |
|
|
|
Geeko
Utente
Citt�: Milano
1043 Messaggi |
Inserito il - 14 maggio 2019 : 11:29:22
|
| Se utilizzi materiali autoclavati (acqua e plastiche) o DNasi-free dovresti essere protetto dalla degradazione. Se hai cambiato i buffer ma erano entrambi vecchi o non autoclavati puoi comunque incorrere nello stesso problema. La contaminazione la deduco dalla banda alta che nel non-digerito ti migra oltre il peso molecolare pi� alto (10kb?) e poi diventa uno smear dopo la digestione. Te ne rimane anche un po' nel pozzetto col vettore. Potrebbe per� anche essere una forma concatenata del vettore. Una volta che riesci a digerirlo correttamente e ottenere bande definite dovresti riuscire a distinguere tra i due casi. |
|
|
|
|
weppa29
Nuovo Arrivato
6 Messaggi |
Inserito il - 18 maggio 2019 : 22:23:35
|
Buonasera a tutti. Finalmente, sono riuscito a digerire correttamente cloni e vettore, defosforilare quest'ultimo, purificare da gel di agarosio, ligare e trasformare.
Oggi ho effettuato una Colony PCR (ho utilizzato 4 colonie per piastra a eccezione dell'ultimo campione) per comprovare che le colonie ottenute contenessero il mio inserto (ho utilizzato primers specifici per il vettore).
Nel penultimo pozzetto ho caricato DNA del vettore come controllo (cerchio in blu).
Il risultato � il seguente...pare che nel caso del primo campione (cerchi rossi) il vettore si sia ricircolarizzato, mentre nel secondo ho differenti prodotti, cio� colonie con solo vettore e colonie con vettore e inserto (cerchi verdi), sbaglio? Grazie!!!!
https://ibb.co/cvTX1pN
|
|
|
|
weppa29
Nuovo Arrivato
6 Messaggi |
Inserito il - 18 maggio 2019 : 22:29:13
|
Un problema che � emerso � anche questo...ho caricato il DNA purificatO da gel per verificare che la purificazione fosse avvenuta correttamente e mi ritrovo questo disastro...
https://ibb.co/ynmrSpZ |
|
|
| |
Discussione |
|


