Struttura secondaria dell'RNAG
1. INTRODUZIONE
1.1 PROCESSO DI INVASIVITÀ E PATOGENESI DI Shigella flexneri
Il genere Shigella appartiene alla famiglia delle Enterobacteriaceae. Comprende batteri Gram-negativi, immobili, anaerobi facoltativi, aventi sia metabolismo respiratorio che fermentativo. Sono patogeni intestinali delluomo e di altri primati nei quali causano la dissenteria bacillare. Nell uomo le lesioni della dissenteria bacillare sono normalmente limitate al retto e allintestino crasso.
Sistematica:
Regno: Procaryotae
Philum: Gracilicutes
Classe: Soctobacteria
Famiglia: Enterobacteriaceae
Genere: Shigella
Specie: S. dysenteriae - S. flexneri - S. boydii - S. sonni
Qualsiasi microrganismo o agente parassita che provoca una malattia infettiva viene definito patogeno e si definisce patogenicità la sua capacità di indurre la malattia.
Con il termine virulenza si intende il grado o lintensità della patogenicità di un certo microrganismo.
Viene valutata sulla base di tre elementi caratteristici del patogeno:
1.Invasività: abilità che un patogeno ha di diffondere nei tessuti adiacenti al sito di infezione o in altri.
2.Infettività: abilità di un patogeno a stabilire un punto focale di infezione nei tessuti di un ospite
3.Potenziale patogeno: grado di capacità del patogeno di determinare sintomi morbosi.
Il processo di invasività della specie S. flexneri è simile a quello di ceppi di Escherichia coli enteroinvasivi (EIEC).
Una volta superata la barriera gastrica, S. flexneri ed EIEC raggiungono il colon dove penetrano la mucosa intestinale attraverso cellule epiteliali specializzate, le cellule M, che rivestono aggregati di tessuto linfoide, presenti lungo tutto il tratto intestinale (placche del Peyer). I batteri vengono fagocitati dalle cellule M nelle quali però, rapidamente, si liberano dal fagosoma che li racchiude, iniziando a moltiplicarsi e a diffondersi sia nelle cellule adiacenti che nella sottomucosa, dove vengono internalizzati dai macrofagi. Invece di essere distrutti dai macrofagi, i batteri si moltiplicano al loro interno e, mediante un meccanismo ancora non del tutto conosciuto, ne inducono lapoptosi, innescando in questo modo una reazione infiammatoria acuta con possibili ulcerazione dellepitelio. Linstaurarsi della reazione infiammatoria da un lato favorisce linvasione delle cellule epiteliali intestinali, in quanto si produce una destabilizzazione delle zone di coesione delle cellule epiteliali (tight junctions); dallaltro stimola la migrazione transepiteliale dei polimorfonucleati che una volta raggiunta la sede dellinfezione contribuiscono a limitarla e/o eliminarla. (Fig.1).
Shigella ed E. coli EIEC inducono la propria internalizzazione nelle cellule epiteliali attraverso la sintesi di proteine specifiche (le proteine Ipa) che vengono esportate da un sistema di secrezione di tipo III, codificato dai geni plasmidici mxi e spa.(Fig.2)
Le proteine Ipa, penetrate nella cellula ospite, attivano sistemi cellulari (signal transduction pathways), che portano a riarrangiamenti consistenti del citoscheletro della cellula i quali, a loro volta, inducono modificazioni della membrana cellulare (ruffling) ed internalizzazione del batterio in un fagosoma. Rapidamente il batterio provoca la lisi del fagosoma liberandosi nel citoplasma dove inizia a moltiplicarsi e muoversi sia allinterno della cellula stessa che verso le cellule adiacenti (Cossart and Sansonetti, 2004).
Figura 1. Invasione batterica della mucosa intestinale.
Destabilizzazione delle tight junctions in seguito alla reazione infiammatoria e migrazione dei leucociti
Figura 2. Durante il contatto con la cellula ospite, lapparato Mxi-Spa inserisce nella membrana cellulare un poro, costituito da IpaB e IpaC. Questo permette il trasferimento degli effettori batterici come IpaA, ma anche lingresso del dominio C-terminale di IpaC nel citosol della cellula. Questultimo, attivando una GTP-asi, induce la polimerizzazione dellactina.
Linterazione tra IpaB e i recettori della superficie cellulare può favorire linserzione del poro (1) o fungere da segnale per il riarrangiamento del citoscheletro (2).
1.2. UN RNA ANTISENSO E COINVOLTO NELLA REGOLAZIONE A LIVELLO TRASCRIZIONALE DEL GENE virG
Lo scopo primario del lavoro di ricerca svolto nellultima decade presso il laboratorio di Genetica Molecolare e dei Microrganismi è stato ed è la comprensione di circuiti di regolazione basati su complicati interplay tra differenti fattori, con particolare attenzione a quei sistemi genetici potenzialmente coinvolti nei processi di patogenicità batterica.
Infatti linvasione di un ospite da parte di un microrganismo patogeno determina una riprogrammazione dellattività trascrizionale e rappresenta quindi un ottimo modello per studiare le risposte adattative globali connesse a variazioni delle condizioni ambientali, che si hanno nel passaggio da una vita extra a una vita intra-ospite.
In questo contesto attualmente il Dr. M. Falconi e collaboratori stanno studiando il gene plasmidico di virulenza virG di Shigella flexneri.
In Shigella i geni di virulenza sono portati su un plasmide (pINV) ad alto peso molecolare (Maurelli and Sansonetti, 1988) che determina il fenotipo invasivo.
Lattivazione dei geni di invasione ipa, mxi e spa si attua con un meccanismo di reazione a cascata basato su un interplay tra differenti regolatori trascrizionali quali VirF, VirB ed H-NS. Inizialmente VirF attiva la trascrizione del gene codificante per lattivatore secondario VirB, che a sua volta è necessario per lespressione dei geni strutturali coinvolti nei processi di invasività (Fig.3) I geni virF e virB sono sottoposti a un complesso meccanismo di regolazione, che risponde a cambiamenti di temperatura, essendo espressi solo a 37 °C ; sono repressi dalla proteina del nucleoide batterico H-NS. (Dorman and Porter, 1998; Falconi et al., 1998; Prosseda et al., 2002).
Figura 3. Cascata regolatoria che determina l'attivazione dei geni di virulenza di Shigella.
VirF attiva la trascrizione del gene codificante per lattivatore secondario VirB, necessario per lespressione dei geni strutturali coinvolti nel processo di invasività. Lespressione della maggior parte dei geni plasmidici di virulenza è modulata dalla temperatura e repressa dalla proteina del nucleoide H-NS (Stella et al., 2005, 2006; Dorman, 2007). Al contrario degli altri geni di invasività, virG non è sottoposto alla cascata regolatoria VirF-VirB, ma è attivato direttamente dalleffettore trascrizionale VirF (Dorman and Porter, 1998; Prosseda et al., 2002).
Il gene strutturale virG di Shigella codifica per una proteina richiesta per linvasione delle cellule epiteliali e per la diffusione intracellulare dei patogeni (Goldberg, 2001). Questa proteina, una tra le più grandi nei batteri (1102 residui aminoacidici), induce la polimerizzazione dellactina ad un polo della cellula, con formazione di una coda di actina che fa muovere il batterio nel citosol della cellula ospite e ne determina la diffusione da una cellula allaltra. ( Fig.4)
La proteina VirG (120 kDa) è costituita da due domini: il dominio α ( a.a. 53-758) contiene il sito che determina lassemblaggio dellactina e protrude dalla superficie del batterio verso lambiente extracellulare. Il dominio β (a.a. 759-1102), è ancorato al foglietto esterno della membrana.
Figura 4. Diffusione del batterio da una cellula all'altra tramite formazione di una coda di actina
Recentemente, nel laboratorio di Genetica Molecolare e dei Microrganismi è stato scoperto che nella regione 5-UTR (Untraslated Region) di virG, è trascritto un RNA antisenso non codificante. Nello specifico lRNA antisenso (denominato RNAG), si origina a partire da una citosina in posizione +120 rispetto al punto di inizio della trascrizione del gene virG ed ha una lunghezza di 450 nucleotidi.
LRNAG è coinvolto nella regolazione a livello trascrizionale del gene virG e appartiene ad unampia classe di piccoli RNA regolatori (sRNA).
Attualmente, sono stati identificati circa 140 piccoli sRNA batterici (50-500 nucleotidi) che non codificano per proteine (ncRNA).
Tali molecole sono state caratterizzate non solo nei batteri, ma anche nei fagi, nei plasmidi e nelle cellule eucariotiche, suggerendo che un simile livello di regolazione è molto diffuso nei processi biologici (Gottesman, 2004).
La funzione di molti sRNA deve ancora essere chiarita. La maggior parte di quelli attualmente caratterizzati, agisce appaiandosi con un secondo RNA, normalmente un RNA messaggero (RNA come antisenso) e cambiandone il comportamento.
I piccoli RNA regolatori possono essere codificati sia in cis, dal filamento opposto dell mRNA bersaglio, oppure da un gene lontano da quello bersaglio. Sono stati descritti diversi meccanismi RNA-RNA mediati, che comportano cambiamenti nel processamento e nella degradazione del messaggero bersaglio e nellefficienza della sua trascrizione e traduzione. (Gottesman, 2004; Storz et al., 2004; Brantl, 2007).
In realtà, è sempre più evidente che gli sRNA, oltre alla regolazione genica in generale, hanno anche un ruolo chiave nel controllare lespressione dei geni di virulenza; inoltre sono coinvolti nella regolazione delle risposte adattative allo stress, importanti per la sopravvivenza del batterio nellospite. Infatti, recenti ricerche hanno identificato gli sRNA in patogeni umani come L. monocytogenes, S. aureus, P. aeruginosa, V. cholerae. (Romby et al., 2006). È interessante notare che in E. coli la maggior parte degli sRNA è presente in ceppi patogeni, confermando lipotesi che essi possano controllare la virulenza.
Recentemente, il lavoro di ricerca svolto nel laboratorio che mi ha ospitato per il periodo dello stage, ha dimostrato che:
1. Lattività del promotore di virG è repressa in presenza dellRNAG ed un prodotto di terminazione del trascritto nativo e stato evidenziato in saggi di trascrizione in vitro.
2. Linibizione trascrizionale è stata osservata anche in vivo usando fusioni cromosomali virG-lacZ quando lRNAG era fornito in trans.
3. Esperimenti di RNA probing con agenti modificanti hanno evidenziato che la struttura della regione leader dellmRNA di virG si modifica in seguito allappaiamento con lRNAG antisenso. In particolare dopo la formazione dellibrido mRNA-RNAG una struttura stem-loop tipica dei terminatori intrinseci (Rho-indipendenti) si origina sul trascritto nascente di virG; tale struttura promuove la prematura terminazione della trascrizione del gene bersaglio.
Ciò rappresenta, se confermato, la prima dimostrazione, in batteri Gram-negativi, di un meccanismo chiamato attenuazione trascrizionale (Brantl, 2002).
2. MATERIALI E METODI
I risultati ottenuti relativi alla regolazione di virG mediata dall RNAG, hanno spinto il gruppo di ricerca del laboratorio di Genetica molecolare, a determinare anche la struttura del suddetto RNA antisenso.. A tale scopo, con laiuto dei collaboratori del Dr. Maurizio Falconi, ho utilizzato la tecnica del probing chimico dellRNA (Fig. XXX).
2.1. MODIFICAZIONE CHIMICA DELLRNAG
LRNA antisenso, estratto e purificato dal gruppo di ricerca del laboratorio in cui ho svolto lo stage, è stato modificato utilizzando due reagenti: dimetil solfato (DMS) e 1-cicloesil-3-(morfolinoetil) carbodiimide meto p-toluene sulfonato (CMCT). Questi reagenti modificano specificamente le basi non appaiate dellRNA; in particolare il DMS modifica le adenine e le citosine; il CMCT modifica le guanine e le uridine. (Holmberg et al., 1994). Sono stati preparati dieci campioni utilizzando la seguente procedura:
Reagenti:
DMS diluito 1:8 in etanolo (3µl DMS in 21µl di etanolo)
CMCT (30 mg/ml): 1,4 mg in 46µl di H2O
RNAG (23 pmoli/µl): 2 pmoli/campione
Condizioni:
Le reazioni sono allestite come schematicamente riportato in tabella.
.Procedura: ( i volumi sono espressi in µl)
Campioni H2O Buffer Sol.A tRNA(1γ/ µl) DMS
DMS: 5x N2 buffer 2 0
1 12.5 4 1.5 2 0
2 12.5 4 1.5 2 0.7
3 11.8 4 1.5 2 1.5
4 11.0 4 1.5 2 2.5
5 10.5 4 1.5 2
CMCT: CMCT buffer CMCT Incubare a 32°C per 20 minuti
6 12.5 4 1.5 2 0
7 12.5 4 1.5 2 0
8 11.5 4 1.5 2 1
9 10.5 4 1.5 2 2
10 8.5 4 1.5 2 4
Inizialmente viene preparata la soluzione A, contenente 1.2 µl di RNAG e 19.2 µl di H2O; questa viene quindi incubata a 90°C per 1 minuto, per denaturare lRNA e poi mantenuta in ghiaccio per 1 minuto. Vengono preparati 10 campioni aggiungendo,alla soluzione A, H2O e 4 µl di tampone. I campioni vengono incubati a 32°C per 10 minuti, trascorsi i quali viene aggiunto il tRNA. (perchè?). Vengono infine aggiunti gli agenti modificanti, in concentrazioni crescenti nei diversi campioni; la modificazione si effettua alla temperatura di 32°C, per 20 minuti con il CMCT e per 5 minuti con il DMS
Al termine della reazione vengono aggiunti 30 µl di acetato di sodio: (0,3 M pH=5,2)
e 150 µl di etanolo al 100 % , per bloccare la reazione e precipitare lRNAG modificato.
I campioni vengono mantenuti a -20°C per 1 ora, centrifugati a 6°C per 12 minuti (13500 rpm), essiccati a 37°C (dopo aver prelevato il sovranatante) e risospesi in 8 µl H2O.
I campioni di controllo vengono trattati allo stesso modo di quelli sperimentali, eccetto per il fatto che ad essi non vengono aggiunti gli agenti modificanti.
Preparazione della soluzione A:
1.2 µl di RNAG (23 pmoli/ µl) + 19.2 µl H2O.
5x N2 DMS buffer: 50 mM sodium cacodylate pH 7.5, 1mM EDTA
CMCT buffer: 50 mM sodium borate, pH 8.0 , 5 mM MgCl2, 100 mM EDTA.
I due reagenti (DMS e CMCT), vengono utilizzati in concentrazioni crescenti nei diversi campioni, permettendo di identificare siti altamente reattivi (modificati a basse concentrazioni di reagente), e siti moderatamente reattivi (modificati a concentrazioni più alte). Con questo metodo le basi non appaiate dellRNAG vengono modificate chimicamente. La trascrittasi inversa utilizzata nel successivo step (primer extension), terminerà la sintesi di cDNA in corrispondenza dei siti modificati.
2.2. MARCATURA DELLOLIGONUCLEOTIDE
Nella primer extension viene utilizzato, come primer, un oligonucleotide sintetico. In particolare per questo esperimento è stato utilizzato un primer denominato virG-100 (si appaia con la regione localizzata 100 basi a monte del sito di inizio della trascrizione di virG), avente la seguente sequenza: 5-AAAGAACTGAAAAGTTGCGGTT-3.
Loligonucleotide è stato sintetizzato senza un gruppo fosfato allestremità 5 e quindi marcato tramite il trasferimento di γ-32P da [γ-32P]ATP, usando lenzima polinucleotide chinasi del batteriofago T4.
La procedura è la seguente:
Reagenti:
primer virG -100: 100 pmoli/ µl
[γ-32P]ATP 10 Ci/l
T4 polinucleotide chinasi (PNK) 5 U/l
Acetato di sodio 3M pH 5.2
Etanolo 100%
10x T4 PNK buffer:
0.5M Tris-Cl (ph 7.6)
0.1M MgCl2
50 mM dithiothreitol
1 mM spermidine HCl
1 mM EDTA ( ph 8.0).
Reazione
[γ-32P]ATP 8 µl
H2O 3.7 µl
T4 PNK 1 µl
10 X T4 PNK buffer 1.5 µl
primer virG -100 0.8 µl
VOLUME FINALE: 15 µl
Dopo una incubazione di 45 minuti a 37°C la reazione viene fermata aggiungendo 30 µl di TE (10 mM Tris + 1 mM EDTA) e lenzima PNK viene denaturato a 65°C per 10 minuti. Per la precipitazione del primer marcato, si aggiungono al campione 5 µl di acetato di sodio 3M e 150 µl di etanolo. La provetta è mantenuta a -20°C per 60 minuti, trascorsi i quali il campione viene centrifugato a 6°C per 12 minuti (13500 rpm) ed il pellet essiccato a 37°C per 10 minuti, dopo aver tolto il sovranatante. Il primer marcato viene quindi risospeso in 40 µl di H2O.
2.3. PRIMER EXTENSION
Con la primer extension è stato possibile sintetizzare un cDNA, utilizzando come stampo per la trascrittasi inversa, lRNAG modificato. Come specificato in precedenza, lenzima termina la reazione di sintesi in corrispondenza delle basi modificate chimicamente dai due reagenti. In tal modo si ottengono frammenti di varia lunghezza che permettono di identificare le zone dellRNAG modificate e, quindi, a singolo filamento.
La primer extension è stata eseguita seguendo questa procedura:
Reagenti:
RNAG (23 pmoli/ µl)
oligo virG-100 marcato con 32P (2 pmoli/ µl)
Trascrittasi inversa (RT) AMV Roche (22 U/ l)
Tampone 5X RT AMV: 250 mM Tris-HCl, 40 mM MgCl2, 150 mM KCl,
5 mM DTT, pH 8,5
2.5 mM dNTP
KOH 3M
Acido acetico 3M
Acetato di sodio 3M
Etanolo 100%
Procedura: ( i volumi sono espressi in µl)
Le reazioni sono allestite come schematicamente riportato in tabella.
Campioni RNAG MixA Incubare a 90°C per 1 minuto, poi mettere in ghiaccio MixB Incubare a 42°C per 45-60 minuti KOH Hydrolisis
Buffer Incubare a 90°C per 3 minuti, poi a 37°C per 2-6 ore A.Acetico NaAc EtOH
1 4 5 5 3 20 6 100 300
2 4 5 5 3 20 6 100 300
3 4 5 5 3 20 6 100 300
4 4 5 5 3 20 6 100 300
5 4 5 5 3 20 6 100 300
6 4 5 5 3 20 6 100 300
7 4 5 5 3 20 6 100 300
8 4 5 5 3 20 6 100 300
9 4 5 5 3 20 6 100 300
10 4 5 5 3 20 6 100 300
Inizialmente viene preparata la mix A, contenente il primer marcato radioattivamente. Questa viene utilizzata per preparare 10 campioni, ai quali viene aggiunto lRNAG modificato; i campioni vengono poi denaturati a 90°C per 1 minuto, quindi posti in ghiaccio per 1 minuto al fine di bloccare la denaturazione. Viene aggiunta la mixB, contenente i nucleotidi necessari per la sintesi di cDNA, e lenzima trascrittasi inversa. La reazione di sintesi procede alla temperatura di 42°C per 45-60 minuti, trascorsi i quali
la reazione viene bloccata aggiungendo KOH e lhydrolysis buffer. I campioni sono prima incubati a 90° per 3 minuti e successivamente a 37°C per 2 ore per idrolizzare lRNA stampo. La reazione è poi neutralizzata con acido acetico ed il cDNA è precipitato con acetato di sodio ed etanolo assoluto. I campioni vengono incubati a -20°C per 60 minuti, centrifugati a 6°C per 12 minuti (13500 rpm). Dopo aver prelevato il sovranatante, il pellet si essicca a 37°C e i campioni vengono infine risospesi in 10 µl di Sample Buffer.
Preparazione della MixA (per 11 campioni):
5 X Roche RT buffer 19.8 µl
32P oligo virG-100 8.6 µl
H2O 26.6 µl
VOLUME FINALE 55 µl
Preparazione della MixB (per 11 campioni):
5 X Roche RT buffer 13.2 µl
2.5 mM dNTP 22 µl
RT AMV Roche 0.88 µl
H2O 29.92 µl
VOLUME FINALE 66 µl
Preparazione dellHydrolysis buffer
1M Tris-HCl pH 7.5 50 µl
0.5M EDTA 15 µl
1% SDS 500 µl
H2O 435 µl
VOLUME FINALE 1585 µl
Preparazione del Sample Buffer
10 mM EDTA 20 µl
Formammide (100 %) 950 µl
H2O 30 µl
xilene cianolo 1 mg
blu di bromofenolo 1 mg
2.4. SEQUENZIAMENTO DELLA REGIONE INTERGENICA VIRA-VIRG
Affinché sia possibile identificare le basi modificate dellRNAG e quindi studiare la struttura della molecola, è necessario avere come riferimento la sequenza del filamento di DNA corrispondente.
I ricercatori del laboratorio che mi ha ospitato per lo stage, dopo aver ottenuto il filamento relativo alla regione intergenica virA- virG, lhanno inserito (clonato) in un plasmide vettore:pGT1127 Questultimo è stato utilizzato per il sequenziamento con il metodo di Sanger, basato sullutilizzo di miscele di deossinucleotidi e dideossinucleotidi. Questi ultimi, mancando del gruppo OH in posizione 3, necessario per linnesco della polimerasi, causano la terminazione della sintesi. La competizione tra lallungamento e la terminazione della catena, dipende dal rapporto tra dNTP e ddNTP in ognuna delle quattro miscele di reazione. Utilizzando i reagenti nella giusta proporzione, è possibile ottenere quattro famiglie di frammenti terminanti rispettivamente a livello delle quattro basi del DNA.
Tutti i frammenti ottenuti differiscono per una sola base di lunghezza e vengono separati in gel di poliacrilammide. Sarà quindi possibile leggere la sequenza dellantisenso per complementarietà.
Vengono preparate 4 miscele di reazione in 4 diversi tubi eppendorf per termociclatore (100 µl), ciascuno dei quali contiene i dNTP e un ddNTP. Il primer virG -100 funge da innesco per la Taq polimerasi, la quale inizia la sintesi, ma si blocca quando incorpora il dideossinucleotide. La reazione avviene nel termociclatore e le condizioni per la PCR sono le seguenti:
DENATURAZIONE iniziale: 95°C X 2 min
DENATURAZIONE 94°C X 20 sec
ANNEALING: 52°C X 20 sec X 30 cicli
ESTENSIONE: 72°C X 25 sec
Estensione finale 72°C x 30 sec
Reagenti:
32P oligo virG-100 (2 pmoli/ µl)
Fidelitaq Polymerase ( 5 U /l)
0.5 mM dATP 302 µg/ml
0.5 mM dCTP 284µg/ml in TE pH=8.0
0.5 mM dGTP 304 µg/ml
0.5 mM dTTP 291 µg/ml
10 mM ddATP 5.7 mg/ml
10 mM ddCTP 5.4mg/ml in TE pH = 8.0
10 mM ddGTP 5.7 mg/ml
10 mM ddTTP 5.5 mg/ml
Procedura:
Mix Seq
10 X Fidelitaq buffer 1.8 µl
32P oligo virG-100 6.2 µl
pGT1127 1 µl (200 ng)
H2O 8.5 µl
Fidelitaq Polymerase 0.5 µl
Figura 6. Ogni tubo eppendorf contiene 2 µl della miscela contenente tutti i dNTP e un ddNTP; vengono poi aggiunti 4 µl della mix seq la cui preparazione è descritta sopra.
I quattro tubi eppendorf vengono inseriti nel termociclatore, programmato rispettando le condizioni sopra descritte. Terminati i 30 cicli, dopo circa unora, i campioni vengono centrifugati brevemente e ad ognuno vengono aggiunti 7 µl di Sample Buffer.
La sequenza così preparata, verrà caricata nel gel di poliacrilammide-urea.
2.5. ELETTROFORESI SU GEL DI POLIACRILAMMIDE-UREA
I frammenti ottenuti dalla primer extension, vengono separati tramite elettroforesi su gel di poliacrilammide-urea. La poliacrilammide, infatti, ha un alto potere risolutivo e permette di separare frammenti che differiscono di una sola base in lunghezza. Lurea è un agente denaturante, che contribuisce a mantenere a singolo filamento il cDNA.
Preparazione del gel:
33.6 g urea
14 ml acrilammide al 40% ( rapporto acrilammide-bis acrilammide = 60:1)
14 ml TBE 5X
H2O fino a 70 ml
La soluzione viene fatta sciogliere sullagitatore, quindi filtrata usando una pompa da vuoto. Vengono quindi aggiunti 35 µl di TEMED (N,N,N,N-tetrametiletilendiamina) e 415 µl di ammonio persolfato (APS) al 10% , rispettivamente iniziatore e catalizzatore della reazione di polimerizzazione dellacrilammide.
I vetri della camera elettroforetica devono essere accuratamente lavati con acqua, NaOH 5M, sapone ed etanolo, per eliminare eventuali residui di precedenti gel o qualsiasi altra impurità presente. Uno dei due vetri viene inoltre trattato con Repel-Silane ( Amersham-Bioscences), sostanza che impedisce al gel di aderire a tale vetro e facilita, quindi, loperazione di rimozione del gel stesso, a corsa terminata.
Una volta pronta la soluzione, questa viene versata nello spazio tra i due vetri e, dopo aver inserito il pettine, lasciata polimerizzare a temperatura ambiente per circa 10 minuti.
Dopo la polimerizzazione si provvede a togliere il pettine e a sistemare i vetri nella camera eletrroforetica. Nei pozzetti vengono caricati 3 µl di ogni campione e 2 µl di sequenza, ottenuta con il metodo di Sanger precedentemente descritto. La corsa elettroforetica procede alla potenza di 38 W per circa due ore, in tampone TBE 1 X.
I coloranti contenuti nel Sample Buffer, fungono da traccianti di corsa: lo xilene cianolo migra come un frammento di 100 basi; il blu di bromofenolo come un frammento di 36 basi. La corsa viene fermata quando il blu raggiunge lanodo. Si procede quindi a rimuovere dalla camera i due vetri, che vengono aperti; viene staccato il gel e poggiato su un foglio di carta 3MM. Questo viene fatto essiccare a 80°C per 1 ora. Il risultato viene infine visualizzato mediante Imager (BioRad), uno strumento collegato al computer che, rilevando la radioattività su supporto solido, fornisce, direttamente sullo schermo, limmagine del gel. ( Fig. 5A )
Con questo metodo ho studiato la struttura relativa alle prime 120 basi dellRNAG, cioè tutta la zona dellantisenso che è complementare al gene bersaglio virG.
3. RISULTATI E DISCUSSIONE
I risultati ottenuti dagli esperimenti sulla regolazione del gene virG, hanno spinto il gruppo di ricerca del Dr. Falconi a determinare anche la struttura dellRNA antisenso.
Quindi, dopo trascrizione e purificazione in vitro, lRNAG è stato trattato con due reagenti specifici per il singolo filamento: il DMS che modifica adenine e citosine, e il CMCT che agisce su guanine e uridine (Ehresmann et al., 1987).
I siti modificati sono stati individuati tramite primer extension, nella quale la trascrittasi inversa sintetizza un cDNA, utilizzando come stampo lRNAG modificato (vedi materiali e metodi). I frammenti sono stati separati elettroforeticamente (Fig. 5A) e i dati sperimentali ottenuti (probing chimico dellRNA) sono stati immessi in un software specifico, MFOLD, (Zuker et al., 1999) che, elaborando tali dati, ha fornito la struttura secondaria delle prime 120 basi dellRNA antisenso ( Fig. 5B ).
Figura 5A. Pattern elettroforetico del probing chimico (DMS e CMCT) dell RNAG.
Figura 5B. Struttura secondaria delle prime 120 basi dell'RNAG, ottenuta rielaborando i dati del pannello A con il programma MFOLD.
In figura 5A, i campioni n. 2 e 3 sono quelli in presenza di differenti concentrazioni di DMS mentre il campione n. 1 rappresenta quello di controllo, ovvero privo dellagente modificante. I campioni n. 5-7 sono invece quelli modificati dal CMCT, di cui il primo (n. 4) è il controllo. Al centro è stata caricata la sequenza (C T A G) dellantisenso, ottenuta con il metodo di Sanger.
Per lanalisi dei dati ottenuti, vanno considerate le zone del gel in cui il campione di controllo non presenta bande; le bande compaiono, invece, nella stessa posizione, nei campioni trattati con il reagente chimico
Ciò indica lavvenuta modificazione in seguito al trattamento chimico, e il conseguente blocco della trascrittasi inversa durante la reazione di primer extension, con formazione di un frammento di cDNA. Le basi modificate sono indicate a lato del gel in figura 5A.. Poiché, come specificato precedentemente, gli agenti modificanti agiscono sulle basi non appaiate, le bande indicano le zone dellantisenso con struttura a singolo filamento. Le parti del gel in cui non è evidente la differenza tra i campioni di controllo e quelli trattati, indicano invece le zone dellRNAG in cui le basi sono appaiate, a formare un duplex di RNA.. Qui infatti la modificazione non è avvenuta, non presentando la molecola di RNA dei siti accessibili al DMS ed al CMCT.
L analisi della struttura rivela la presenza di tre stem-loop nelle posizioni 3-14, 24-42 e 72-105. Quello principale (pos. 72-105) è caratterizzato da due loop uno apicale ed uno interno. Inoltre è da notare unampia zona sottoposta alla modificazione da parte di entrambi i reagenti, che va dall adenina in posizione 43 a quella in posizione 73. Ciò indica che tale zona non è strutturata e si presenta a singolo filamento.
Il completamento (previsto in tempi brevi) della struttura secondaria di questo RNA di 450 nucleotidi è attualmente in progresso nel laboratorio di Genetica Molecolare e dei Microrganismi. Tale lavoro potrebbe fornire delle preziose indicazioni sul modo di azione di questi piccoli RNA, presenti sia nei batteri che nelle cellule eucariotiche, nellambito dei processi di regolazione dellespressione genica.
4. CONCLUSIONI
Ho svolto il mio stage presso il laboratorio di Genetica Molecolare e dei Microrganismi, essendo particolarmente interessata agli aspetti molecolari della genetica.
Durante il periodo dello stage ho avuto modo, in una prima fase, di osservare la messa a punto di molte metodiche sperimentali e, successivamente, di mettere alla prova la mia manualità applicando le tecniche sopra descritte.
Ciò è stato di grande utilità per me, poiché mi ha permesso di acquisire sicurezza, attenzione e precisione: tutti i protocolli, infatti, hanno previsto lutilizzo di sostanze radioattive anche se in quantità molto piccole (pochi µCi), e quindi la necessità di pipettare dietro appositi schermi di protezione, ponendo estrema attenzione alle possibili contaminazioni.
È stato inoltre molto interessante laver preso confidenza con le metodologie descritte: si tratta infatti di protocolli molto particolari, che non ho mai avuto modo di applicare nel corso dei miei studi e che difficilmente avrei potuto applicare svolgendo il mio stage in unaltra azienda.
A questo proposito ringrazio il Dr. Maurizio Falconi e tutti i suoi collaboratori (Dr. M. Giangrossi, A. Brandi e G. C. Tran), che mi hanno sempre attentamente seguito chiarendo ogni mio dubbio o quesito.
5. REFERENZE
Lansing M. Prescott, John P. Harley, Donald A. Klein: Microbiologia
I edizione ZANICHELLI
Brantl S. ( 2002). Antisense-RNA regulation and RNA interference. Biochim. Biophys Acta, 1575, 15-25.
Brantl, S. ( 2007). Regulatory mechanisms employed by cis-encoded antisense RNAs. Curr. Opin. Microbiol. 10, 102-109.
Cossart, P., and Sansonetti, PJ. (2004). Bacterial invasion: the paradigms of enteroinvasive pathogens Science 304, 242-248.
Dorman, C.J., and Porter, M.E. (1998). The Shigella virulence gene regulatory cascade: a paradigm of bacterial gene control mechanisms. Mol. Microbiol. 29, 677-684.
Dorman, C.J. (2007). H-NS, the genome sentinel. Nature Rev. 5, 157-161.
Ehresmann, C., Baudin, F., Mongel, M., Romby, P., Ebel, J P., and Ehresmann, B. (1987). Probing the structure of RNAs in solution. Nucleic Acids Res. 15, 91099128.
Falconi, M., Colonna, B., Prosseda, G., Micheli, G., and Gualerzi C.O. (1998). Thermoregulation of Shigella and Escherichia coli EIEC pathogenicity. A temperature-dependent structural transition of DNA modulates accessibility of promoter to trascriptional repressor H-NS. EMBO J.:7033-7043.
Goldberg, M.B. (2001). Actinbased motility of intracellular microbial pathogen. Microbiol. Mol. Biol. Rev.65, 595-626.
Gottesman, S. (2004). The small RNA regulators of Escherichia coli: roles and mechanisms. Annu. Rev. Microbiol. 58, 303-328.
Holmberg, L., Melander, Y., and Nygård, O. (1994) Probing the structure of mouse Ehrlich ascites cell 5.8,18S and 28S ribosomal RNA in situ Nucleic Acids Res. 22, 13741382.
Maurelli,A.T. and Sansonetti,P.J. (1988) Identification of a chromosomal gene controlling temperature regulated expression of Shigella virulence. Proc. Natl Acad. Sci. USA, 85, 28202824.
Prosseda, G., Falconi, M., Nicoletti, M., Casalino, M., Micheli, G., and Colonna, B. (2002). Histone-like proteins and Shigella invasivity regulon. Res. Microbiol. 153, 461-468.
Romby, P., Vandenesch. F., and Wagner, E.G.H. (2006). The role of RNAs in regulation of virulence-gene. Curr. Opin. Microbiol. 9, 1-8.
Sambrook, J., and Russell, D.W. (2001). Molecular Cloning. A Laboratory Manual. Cold Spring Harbor, NY: CSHL Press.
Stella, S., Falconi, M., Lammi, M., Gualerzi, C.O., and Pon C.L. (2006) Environmental control of the in vivo oligomerization of nucleoid protein H-NS. J. Mol. Biol. 355, 169-174.
Stella, S., Spurio, R., Falconi, M., Pon, C.L., and Gualerzi, C.O. (2005) Nature and mechanism of the in vivo oligomerizzation of nucleoid protein H-NS. EMBO J. 24, 2896-2905.
Storz, G., Opdyke, J.A., and Zhang, A. (2004). Controlling mRNA stability and translation with small, noncoding RNAs. Curr. Opin. in Microbiol. 7, 140-144.
Zuker, M., Mathews, D.H., and Turner, D.H. (1999). Algorithms and thermodynamics for RNA secondary structure prediction: a practical guide in RNA biochemistry and biotechnology. In NATO ASI Series. Barciszewski, J., and Clark, B.F.C. (eds). Dordrecht, the Netherlands: Kluwer Academic Publishers.
Inoltra:
 |
Vota: 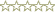
|
|



