Proteina Prionica: Struttura e fisiologia

La proteina prionica cellulare di membrana (PrP
C) è codificata dal gene PRNP situato sul braccio corto del cromosoma 20 dell'uomo e, sebbene sia composto da due esoni, l'intero "open reading frame (ORF)" è localizzato solo nel secondo esone.
La proteina PrP
C è composta da 253 amminoacidi, ha un peso complessivo di 33 - 35 kDa e viene espressa in maniera ubiquitaria soprattutto a livello del sistema nervoso.
A livello post - traduzionale avvengono tre modificazioni principali che sono:
- N - glicosilazioni che danno origine a tre isoforme: monoglicosilata, diglicosilata e non glicosilata
- la rimozione proteolitica di un peptide di 22 amminoacidi all'estremità N - terminale
- la sostituzione del C - terminale con un complesso glicosil - fosfatidilinositolo (GPI) per l' ancoraggio della proteina alla membrana citoplasmatica.
Il ruolo della PrP non è ancora stato chiarito, anche se sono state proposte diverse ipotesi sulla sua funzione:
- Omeostasi del rame
- Interazione con laminina -> Adesione cellulare/sviluppo di neuriti
- Regolazione del flusso di ioni Ca++ -> Trasmissione sinaptica
- Espressa per lo più a livello del sistema nervoso -> Trasduzione del segnale nervoso.
- Azione anti ossidante
- Regolazione del ritmo circadiano
- Azione anti apoptotica
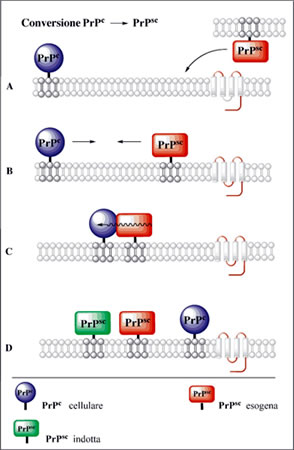
La PrP
C diviene pericolosa in seguito ad un mutamento conformazionale che può essere indotto da un prione infettante esogeno. Il prione esogeno si inserisce nella membrana plasmatica, dove è presente la proteina prionica cellulare, e venendone in contatto diretto ne causa la conversione con un meccanismo ancora sconosciuto (Fig.1). Una volta formata, la PrP
SC, induce cambiamenti conformazionali nelle altre PrP
C attraverso un meccanismo autocatalitico fino a quando non viene raggiunta una concentrazione critica che porta all'aggregazione delle proteine stesse (fibre amiloidi) e al danneggiamento del tessuto nervoso. È stato osservato che può anche avvenire una mutazione genetica spontanea che porta alla formazione di PrP
SC che agisce su altre PrP
C con una reazione a catena.
La PrP
SC presenta anch'essa delle caratteristiche peculiari che sono:
- assenza di potere immunogeno ( non si sviluppano anticorpi)
- assenza di potere infiammatorio (viene riconosciuta come self dall'organismo)
- grande resistenza al calore secco e alle radiazioni UV
- sensibilità (ipoclorito di sodio per 30 min./autoclave 134°- 138°C/NaOH 1N O/N)
I prioni sono composti per la maggior parte da proteina prionica PrP
SC. Anche se la formazione della PrP
SC dalla PrP
C è un processo post traduzionale nessuna modificazione chimica è stata identificata. Ciò suggerisce che la sintesi di PrP
SC sia dovuta ad un cambiamento conformazionale. Per verificare tale possibilità è stata purificata sia PrP
C che PrP
SC usando procedure non denaturanti determinando la struttura secondaria di ognuna.
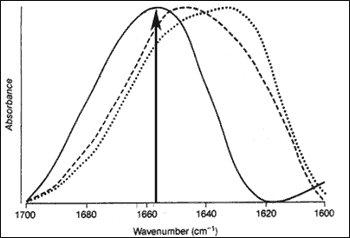
Lo spettro ricavato dall'analisi FT IR della proteina purificata ha dimostrato che PrP
C , presenta un picco simmetrico con un massimo a 1653 cm-1 (Fig.2).
Tale spettro è caratteristico delle proteine con un elevato contenuto in α elica (42%) e non in foglietto β (3%) (Fig.3), queste proporzioni sono state poi confermate tramite DC (Dicroismo Circolare);
al contrario PrP presenta un elevato contenuto di foglietti - β (43%) e un basso contenuto in α - eliche (30%) (Fig.4). Il dicroismo circolare non può essere effettuato per la PrP
SC in quanto è insolubile .
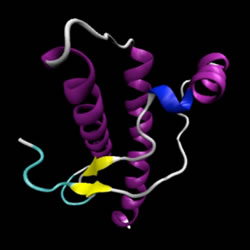
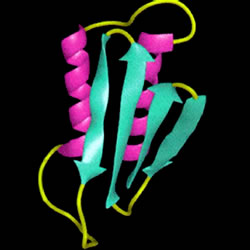
L'analisi di dinamica molecolare ha suggerito che la PrP
C può ripiegarsi in un "bundle" di tre o quattro eliche e in una piccola porzione di foglietto-β. Mentre nella PrP
SC si presentano una serie di foglietti - β disposti in parallelo.
Da diversi studi è stato rilevato che il frammento, derivato da una parziale proteolisi della PrP
SC e responsabile della formazione di fibre amiloidi è il PrP
SC 27-30, costituito da 142 amminoacidi, che presenta un contenuto del 50% di foglietti - β pieghettati a bassa frequenza, ciò riflette una associazione intermolecolare che caratterizza le fibre amiloidi.
Nella produzione di PrP
SC oltre a PrP
C sono necessarie altre proteine che presumibilmente catalizzano l'unfolding delle α - eliche in PrP
C e il refolding di questi domini in β - sheet. Queste proteine ausiliarie sembrano avere proprietà in comune con le chaperonine. La moltiplicazione dell'agente patogeno procede tramite un processo di cristallizzazione coinvolgendo la formazione amiloide di PrP.
Le PrP
SC perdono la loro funzione e tendono ad accumularsi nella sostanza grigia formando le placche amiloidi.
Le principali malattie causate da prioni sono quindi a carico del sistema nervoso, sono neurodegenerative e vengono classificate in:
Encefalopatie Spongiformi Trasmissibili (TSE)
UOMO
- malattia di Creutzfeldt Jakob (MCJ)
- sindrome di Gerstmann Straussler Scheinker (GSS)
- insonnia familiare fatale (IFF).
ANIMALI
- scrapie della pecora e della capra
- encefalite spongiforme del bovino (BSE)
- encefalite trasmissibile del visone (TME)
- malattia cronica devastante del cervo (CWD)
Bibliografia
- Simone Barocci. Seminario sulle malattie da prioni (Facoltà di Scienze MM.FF.NN dell'Università Politecnica delle Marche. Ancona,1 Dicembre 2005)
- Loredana Ingrosso, Maurizio Pocchiari, ISS. Sussidio tecnico scientifico al seminario sulle encefalopatie trasmissibili (Facoltà di Medicina e Chirurgia dell'Università degli Studi di Perugia, 28 Marzo 2006).
- CNR Center of studies on molecular biology. From good-protein to bad- protein. Prion role in human brain diseas (1998).
- Stanley B. Prusiner, Keh- Ming Pan, Michael Baldwin, Jack Nguyen, Maria Gasset, Ana Serban, Darlene Groth, Ingrid Mehlorn, Ziwei Huang, Robert J. Fletterick, Fred E. Cohen. Conversion of α-helices into β-sheets features in the formation of the scrapie prion proteins. Biochemistry (10 Agosto 2003).
- S. Sorbi, F. Massaro. Neurodegenerazione delle demenze e amiloidosi. Neurol Sci (2004).
- S. Sorbi, S. Bagnoli, E. Cellini, P. Forleo. Patologie neurologiche da misfolding. Neurol Sci (2004).
- Christopher M. Dobson. Protein folding and misfolding. Nature (18/25 Dicembre 2003).
- Jeremy M. Berg, John L. Tymoczko, Lubert Stayer, 2003. Biochimica, 5th ed. Zanichelli.
Immagini
- Fig. [2] da Stanley B. Prusiner et all. Conversion of α-helices into β-sheets features in the formation of the scrapie prion proteins. Biochemistry (10 Agosto 2003).
- Fig. [8] da http://www.udel.edu/chem/bahnson/chem645/websites/Janas
- Fig.[1] realizzata con ChemBioDraw da http://www-ermm.cbcu.cam.ac.uk/02005446h.htm
- Fig.[3] realizzata con Visual Molecular Dynamics VMD da relativo .pdb
Inoltra:
 |
Vota: 
|
|